RNA-seq概述
转录组测序原理与实验流程
王诗翔 副教授
中南大学生物医学信息系
中南大学生物医学信息系
2026-03-21
课程大纲
本讲内容
- RNA-seq技术简介
- 实验流程与文库制备
- 测序原理与平台
- 实验设计原则
- 批次效应与控制
- 数据质控指标
- 测序深度与重复数
- 常见问题
第1部分:RNA-seq技术简介
1.1 什么是RNA-seq?
RNA-seq(RNA sequencing) 是利用高通量测序技术对转录组进行定量和定性分析的方法
RNA-seq的核心能力
- 定量分析 — 测定每个基因的转录本丰度
- 差异分析 — 比较不同条件下基因表达变化
- 新转录本发现 — 鉴定新的基因和剪接异构体
- …
1.2 RNA-seq vs 芯片技术
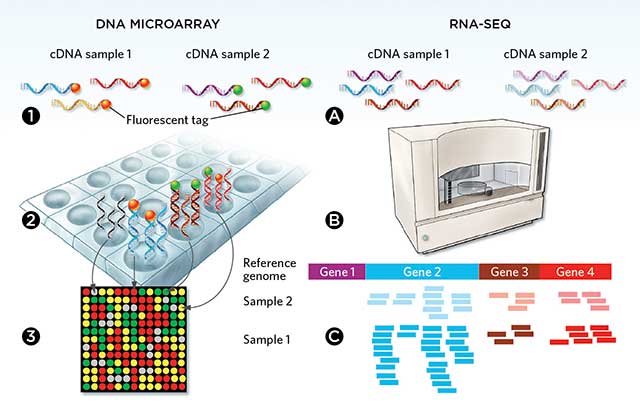
| 特性 | RNA-seq | 芯片(Microarray) |
|---|---|---|
| 检测范围 | 全转录组(已知+未知) | 预设探针(已知基因) |
| 灵敏度 | 高,可检测低表达基因 | 中等,受背景噪音影响 |
| 动态范围 | 宽(>10^4) | 窄(~10^2) |
| 发现能力 | 新转录本、融合基因 | 限于已知序列 |
| 成本 | 逐渐降低 | 相对较低 |
结论:RNA-seq已成为转录组研究的标准方法
1.3 RNA-seq生物医学应用
研究领域
- 🧬 肿瘤研究 — 癌症驱动基因、分型标志物
- 💊 药物研发 — 药物反应预测、靶点发现
- 🧫 发育生物学 — 时空表达模式分析
- 🦠 免疫学 — 免疫细胞活化状态评估
临床转化
- 疾病分子分型
- 预后标志物发现
- 治疗靶点筛选
- 精准医疗指导
第2部分:实验流程
2.1 整体流程图
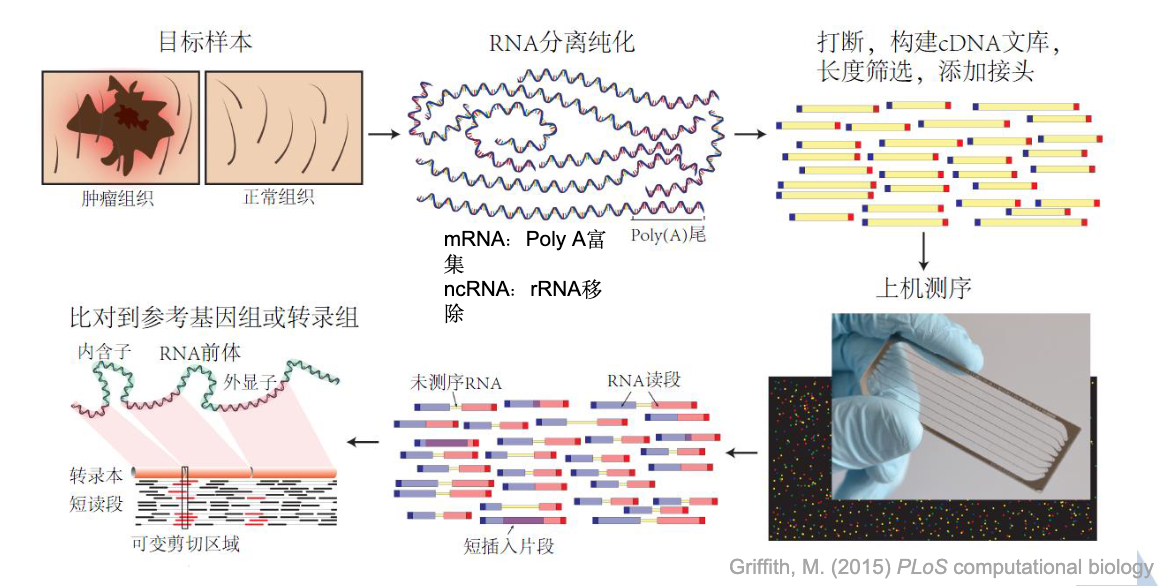
关键质控点:
- RNA完整性(RIN > 7)
- 文库浓度和片段大小
- 测序质量(Q30 > 85%)
2.2 RNA提取与质控
关键指标
| 指标 | 要求 | 说明 |
|---|---|---|
| RIN值 | > 7 | RNA完整性,Bioanalyzer检测 |
| 浓度 | >100 ng/μL | NanoDrop或Qubit检测 |
| 纯度 | A260/A280 = 1.8-2.1 | 蛋白污染检测 |
警告
注意:RNA易降解,提取后应立即进行后续步骤或-80°C保存
2.3 mRNA富集策略
poly-A选择
适用:真核生物、完整RNA样本
优点:
- 富集成熟mRNA
- 数据利用率高
- rRNA污染低
缺点:
- 丢失非编码RNA
- 无poly-A的转录本丢失
- 不适合降解样本
rRNA去除
适用:原核生物、降解样本
优点:
- 保留非编码RNA
- 更全面的转录组
- 适合FFPE样本
缺点:
- rRNA残留可能较高
- 数据复杂度增加
第3部分:测序原理
3.1 Illumina测序(边合成边测序)
原理步骤
- 文库变性 — 双链DNA变性为单链
- 杂交 — 与flow cell上的接头序列结合
- 桥式PCR扩增 — 形成簇(cluster)
- 测序引物结合
- 循环测序 — 加入荧光标记的dNTP
- 图像采集 — 读取每个碱基的荧光信号
3.2 测序模式选择
| 模式 | read长度 | 应用场景 | 成本 |
|---|---|---|---|
| SE50 | 50 bp单端 | 基因表达定量 | 低 |
| SE75 | 75 bp单端 | 常规表达分析 | 中 |
| PE100 | 100 bp双端 | 剪接分析、新转录本 | 高 |
| PE150 | 150 bp双端 | 复杂转录组、融合基因 | 最高 |
推荐:一般表达分析用PE100-150
3.3 测序深度与重复数
推荐测序深度
| 分析类型 | reads/样本 | 说明 |
|---|---|---|
| 基因表达定量 | 10-30 M | 标准差异分析 |
| 差异剪接 | 50-100 M | 需要更高覆盖度 |
| 融合基因检测 | >100 M | 低丰度事件检测 |
| 单细胞测序 | 50K-500K/细胞 | 依细胞类型而定 |
生物学重复数
- n=3:最低要求(细胞系)
- n=4-5:推荐(人临床样本)
- n≥6:高异质性样本
原则:生物学重复 > 测序深度
第4部分:实验设计
4.1 实验设计基本原则
1. 对照组设置 — 必须设置对照组
2. 随机化 — 样本处理顺序随机
3. 平衡设计 — 各组样本数相等
4. 盲法 — 减少操作偏差
5. 详细记录 — 批次、日期、操作者重要
关键:实验设计决定分析的上限,设计缺陷无法通过分析弥补
4.2 常见实验设计类型
两两比较(最常见)
对照组 (n=3) vs 处理组 (n=3)
↓ ↓
提取RNA 提取RNA
↓ ↓
建库测序 建库测序
↓ ↓
←—— 差异分析 ——→配对设计
同一患者:治疗前 (n=20) vs 治疗后 (n=20)
↓
配对分析:控制个体差异,提高统计效能第5部分:批次效应
5.1 什么是批次效应?
批次效应(Batch Effect):由非生物学因素引起的系统性差异
主要来源
| 来源 | 影响程度 | 示例 |
|---|---|---|
| 操作者 | 高 | 不同人员操作习惯差异 |
| 时间 | 高 | 不同日期提取RNA |
| 试剂批次 | 中 | 不同批次试剂性能差异 |
| 仪器 | 中 | 不同测序仪、flow cell |
5.2 批次效应识别
可视化方法
- PCA — 批次是否聚类在一起
- 热图聚类 — 样本是否按批次聚类
- UMAP/t-SNE — 高维数据可视化
异常表现
❌ 错误:样本按批次聚类,而非生物学分组
✅ 正确:样本按生物学条件聚类5.3 批次效应控制
设计阶段控制(首选)
✅ 推荐:块设计(Block Design)
Day 1: 对照1-3 + 处理1-3
Day 2: 对照4-6 + 处理4-6
❌ 避免:
Day 1: 所有对照组
Day 2: 所有处理组分析阶段校正
| 方法 | R包 | 适用场景 |
|---|---|---|
| ComBat | sva | 已知批次变量 |
| ComBat-seq | sva | RNA-seq计数数据 |
| 纳入设计公式 | DESeq2 | 批次作为协变量 |
第6部分:数据质控
6.1 测序质量分数(Phred Score)
Q = -10 × log10(P) (P为错误概率)| Q值 | 准确率 | 错误率 | 评价 |
|---|---|---|---|
| 30 | 99.9% | 0.1% | 优秀 |
| 20 | 99% | 1% | 可接受 |
| <20 | <99% | >1% | 差 |
要求:Q30 > 85%
6.2 比对率指标
| 指标 | 可接受 | 良好 | 优秀 |
|---|---|---|---|
| 总比对率 | >70% | >85% | >90% |
| 唯一比对率 | >60% | >75% | >85% |
| rRNA比例 | <10% | <5% | <2% |
警告
低比对率可能原因:
- 接头未去除干净
- 样本污染
- 参考基因组选择错误
第7部分:常见问题
7.1 RNA-seq常见问题
Q1: 选择poly-A还是rRNA去除?
- 完整RNA+真核生物 → poly-A
- 降解样本/原核生物 → rRNA去除
Q2: 单端还是双端测序?
- 仅表达定量 → SE75
- 剪接分析/新转录本 → PE100-150
Q3: 如何判断测序深度是否足够?
- 基因检出数(通常>10,000)
- 饱和曲线分析
- 样本间相关性
7.2 实验设计检查清单
总结
RNA-seq实验要点
- 技术优势 — 全转录组覆盖、高灵敏度、发现新转录本
- 实验流程 — RNA提取 → mRNA富集 → 片段化 → cDNA合成 → 接头连接 → 测序
- 测序参数 — PE100-150用于剪接分析,10-30M reads/样本,n≥3重复
- 实验设计 — 对照组、随机化、平衡设计、详细metadata记录
- 批次效应 — 设计阶段控制优于分析阶段校正
谢谢!
联系方式
- 📧 wangshx@csu.edu.cn
- 🌐 https://wanglabcsu.github.io/
- 🐙 https://github.com/WangLabCSU
延伸阅读
- Wang Z, et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009
- Conesa A, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016
- Leek JT, et al. Tackling the widespread and critical impact of batch effects. Nat Rev Genet. 2010
![]()
RNA-seq概述 | 中南大学