speed dist
Min. : 4.0 Min. : 2.00
1st Qu.:12.0 1st Qu.: 26.00
Median :15.0 Median : 36.00
Mean :15.4 Mean : 42.98
3rd Qu.:19.0 3rd Qu.: 56.00
Max. :25.0 Max. :120.00 R Markdown与可重复研究
R语言在生物信息学中的应用
王诗翔 副教授
中南大学生物医学信息系
中南大学生物医学信息系
2025-01-01
课程大纲
本讲内容
- 什么是R Markdown
- Markdown基本语法
- R Markdown文档结构
- 代码块选项
- Quarto:下一代工作流
- 可重复性研究最佳实践
第1部分:什么是R Markdown?
1.1 传统科研流程的痛点
❌ 传统方式
- Excel + Word + PPT 分离
- 数据更新后手动重绘图表
- 代码和文档不同步
- 难以复现分析过程
- 合作者看不懂分析步骤
✅ R Markdown方式
- 代码 + 文档 一体化
- 数据更新自动刷新图表
- 一处修改,全局更新
- 完全可重复的分析
- 生成HTML / PDF / Word多格式
💡 R Markdown实现了文学编程(Literate Programming)的理念
1.2 R Markdown的核心价值
.Rmd文件 → knitr执行R代码 → Markdown → pandoc渲染 → HTML/PDF/Word
↑______________________________________________|
(数据更新后重新knit即可)三大核心优势
- 动态报告 — 数据变化,报告自动更新
- 可重复性 — 他人可完全复现分析过程
- 多格式输出 — 一份源码,生成多种格式
参考:Yihui Xie 的 R Markdown: The Definitive Guide
https://bookdown.org/yihui/rmarkdown/
第2部分:Markdown基本语法
2.1 什么是Markdown?
Markdown是轻量级标记语言,用纯文本格式编写,一键转换为格式化文档
设计哲学
“让格式语法尽可能可读” — John Gruber(Markdown创始人)
优势
- ✅ 纯文本,版本控制友好(Git追踪变化)
- ✅ 易读易写,专注内容
- ✅ 一键转换为 HTML / PDF / Word / 幻灯片
2.2 标题与文本格式
粗体文字 斜体文字 删除线
这是一段引用文字(blockquote)
2.3 列表与链接
- 无序列表项目一
- 子项目(缩进2个空格)
- 无序列表项目二

2.4 表格语法
| 基因 | 表达量 | 显著性 | 调控方向 |
|---|---|---|---|
| TP53 | 12.5 | *** | 上调 |
| KRAS | 8.3 | * | 上调 |
| BRCA1 | 3.1 | ns | 下调 |
2.5 代码与公式
行内代码与代码块
这里只会标记代码,不会像 RMarkdown 里代码可以执行
2.5 代码与公式
LaTeX数学公式
第3部分:R Markdown文档结构
3.1 R Markdown的组成
在RStudio中创建:File → New File → R Markdown
三大组成: 1. YAML元数据(---包围的头部) 2. Markdown文本(叙述性内容) 3. R代码块(以```{r} 开始)
3.2 YAML元数据配置
3.3 代码块基本语法
R Markdown传统语法
3.3 代码块基本语法
常用选项速查
| 选项 | 说明 | 默认值 |
|---|---|---|
echo |
是否显示代码 | TRUE |
eval |
是否执行代码 | TRUE |
results |
输出处理方式 | 'markup' |
warning |
是否显示警告 | TRUE |
message |
是否显示消息 | TRUE |
fig.width |
图宽(英寸) | 7 |
fig.height |
图高(英寸) | 5 |
fig.cap |
图片标题 | NULL |
3.4 代码块演示
① 只显示结果(隐藏代码)
3.4 代码块演示
② 只显示代码(不执行)
③ 隐藏一切(后台运行)
3.5 内联代码(Inline Code)
在文本中嵌入R计算结果,实现动态更新
本次分析共 50 个样本, 平均速度 15.4 mph, 中位刹车距离 36 英尺。
生信应用场景
- 正文中自动更新样本量、P值
- 确保正文与表格一致(避免手动填错)
- 比较结果描述随数据自动调整
3.6 图表输出
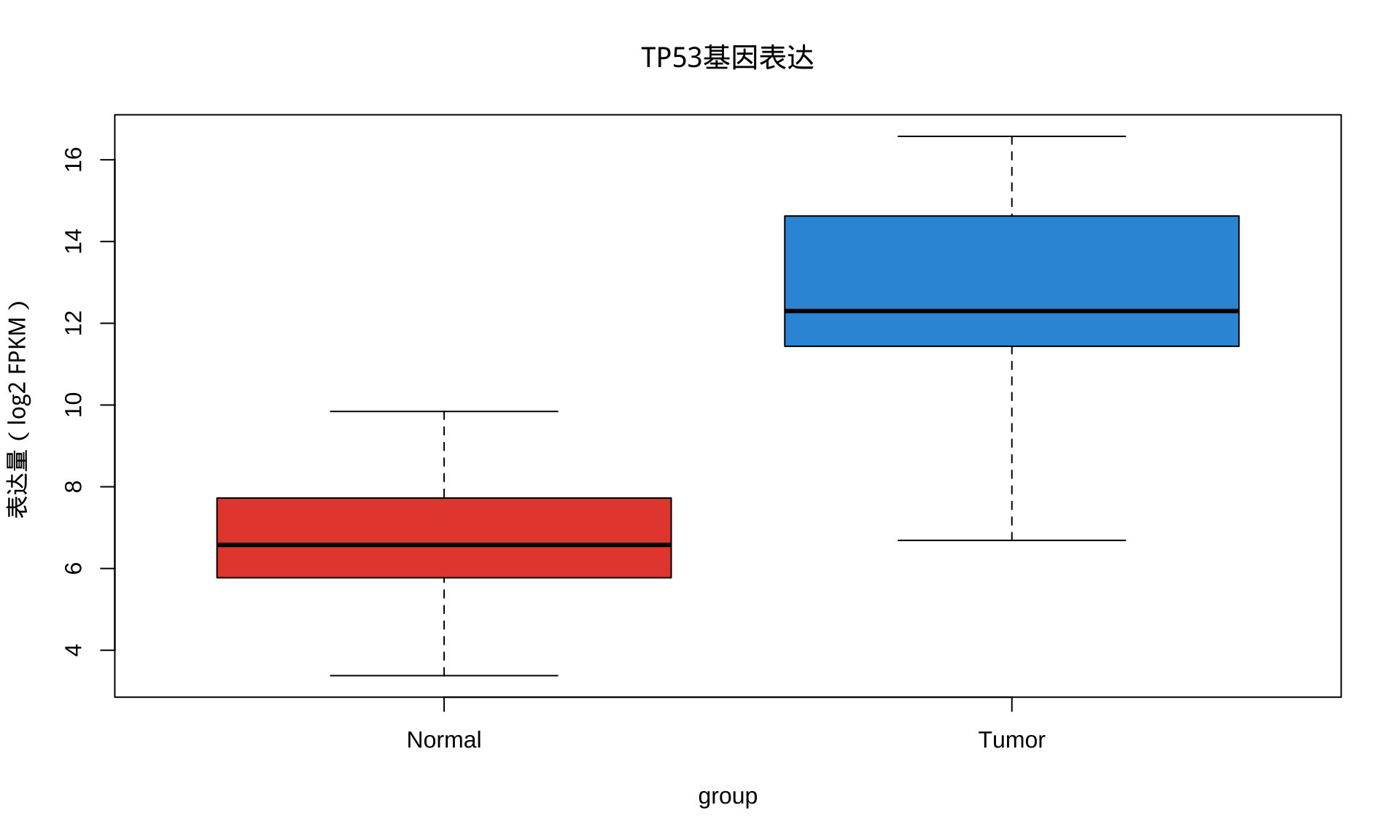
3.7 ggplot2可视化
library(ggplot2)
ggplot(df, aes(x = group, y = expression, fill = group)) +
geom_boxplot(alpha = 0.7, outlier.shape = NA) +
geom_jitter(width = 0.2, size = 2, alpha = 0.6) +
scale_fill_manual(values = c("Normal" = "#3498DB", "Tumor" = "#E74C3C")) +
labs(title = "TP53基因表达:肿瘤 vs 正常",
x = "样本分组", y = "表达量(log2 FPKM)") +
theme_minimal(base_size = 14) +
theme(legend.position = "none")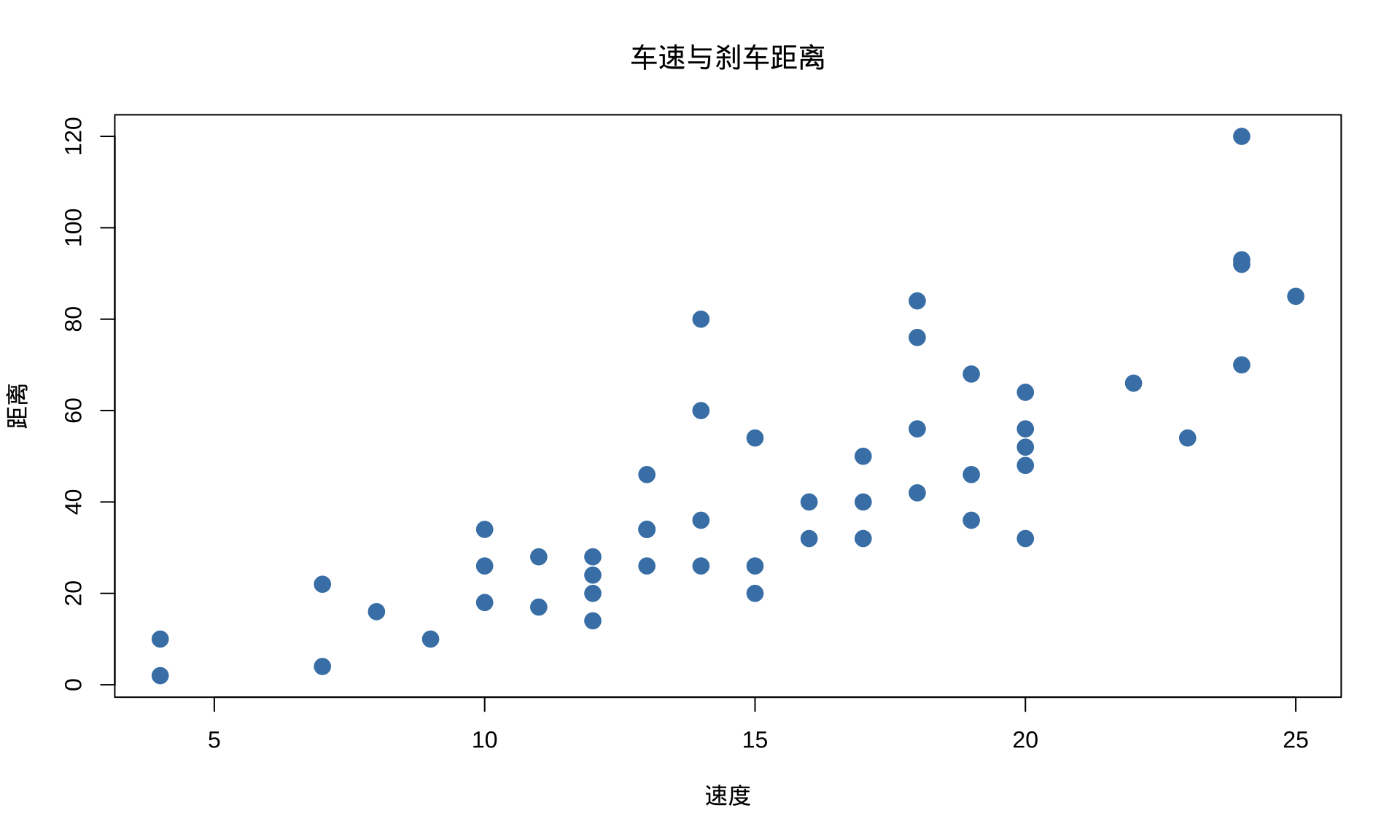
3.8 表格输出
使用 knitr::kable
| 基因 | log2 FC | P值 | 校正P值 |
|---|---|---|---|
| TP53 | 1.93 | 0.0012 | 0.0048 |
| KRAS | -0.82 | 0.0435 | 0.1305 |
| BRCA1 | 2.15 | 0.0003 | 0.0012 |
| MYC | 1.47 | 0.0089 | 0.0267 |
3.9 参数化报告
定义参数(在YAML中)
在代码块中使用参数
第4部分:Quarto:下一代工作流
4.1 什么是Quarto?
Quarto (https://quarto.org/)是下一代开源科学出版系统,统一支持R、Python、Julia等多语言
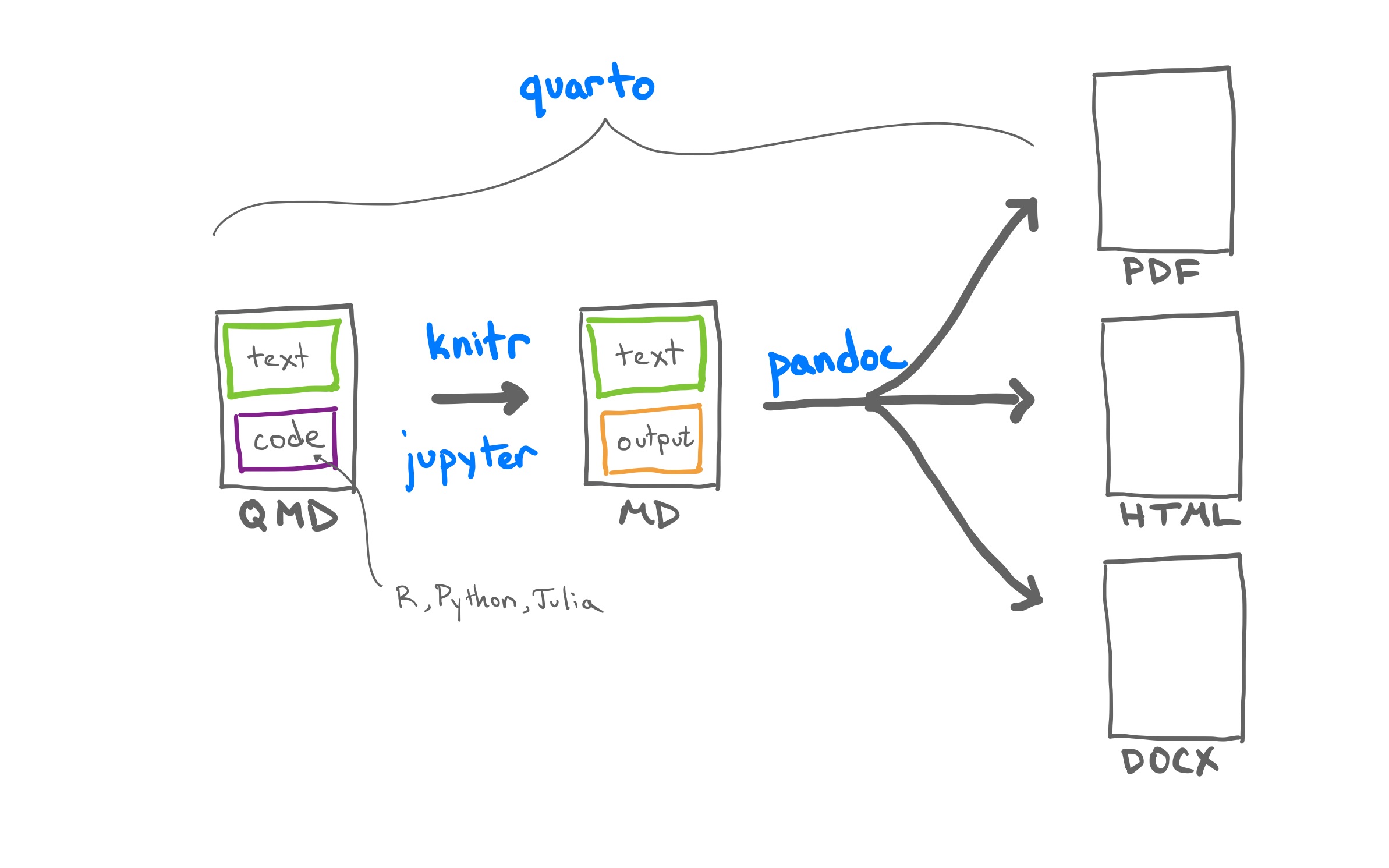
4.1 什么是Quarto?
与R Markdown的关系
R Markdown → Quarto
R专属 → 多语言统一平台- R Markdown仍然受支持
- Quarto是面向未来的选择
- 官网:https://quarto.org/
核心优势
- 🌐 多语言:R + Python + Julia + Observable
- 📱 响应式设计:移动端友好
- 🎨 现代外观:更美观的默认样式
- 🔗 原生交叉引用:无需bookdown
- 📚 多格式:网站/书籍/幻灯片/论文
4.2 Quarto vs R Markdown
| 功能 | R Markdown | Quarto |
|---|---|---|
| 代码块选项 | {r, echo=FALSE} |
#\| echo: false |
| 图宽 | fig.width=8 |
#\| fig-width: 8 |
| 图标题 | fig.cap="..." |
#\| fig-cap: "..." |
| 交叉引用 | 需bookdown | 原生支持 |
| 多语言 | 主要是R | R/Python/Julia |
| 网站 | blogdown/pkgdown | 原生支持 |
| 幻灯片 | xaringan/ioslides | revealjs原生支持 |
4.3 Quarto代码块语法
Quarto风格(推荐)
交叉引用
4.4 Quarto多语言支持
第5部分:可重复性研究最佳实践
5.1 可重复性研究原则
为什么重要?
“科学的进步建立在可重复验证的基础上”
5.2 代码书写规范
命名规范
函数文档(roxygen2风格)
5.3 生信分析报告模板
标准报告结构
5.4 输出格式选择
根据场景选择格式
| 场景 | 推荐格式 | 配置 |
|---|---|---|
| 实验室内部汇报 | HTML | output: html_document |
| 论文附件/投稿 | output: pdf_document |
|
| 与非R用户协作 | Word | output: word_document |
| 展示汇报 | Reveal.js幻灯片 | format: revealjs |
| 在线课程/教材 | Quarto网站 | project: website |
课程总结
本讲要点回顾
Markdown
- 轻量级标记语言,专注内容
- 标题(
#)、粗体(**)、斜体(*) - 列表、表格、链接、公式
R Markdown
- 三要素:YAML + Markdown + 代码块
- 代码块选项控制输出
- 内联代码实现动态报告
- 参数化报告批量生成
Quarto
- 下一代科学出版系统
- 多语言支持(R/Python/Julia)
- 原生交叉引用和多种格式
最佳实践
- renv管理依赖环境
- 规范的项目目录结构
- sessionInfo()记录环境
- 代码命名清晰规范
课程作业
基础任务(必做)
- 用R Markdown创建一份TP53基因表达分析报告,包含:
- 研究背景(Markdown文本)
- 数据生成(R代码块)
- 箱线图(ggplot2)
- 差异分析结果(knitr::kable表格)
- 内联代码描述结果
进阶任务(选做)
- 将R Markdown报告转换为Quarto格式(使用
#|选项) - 创建参数化报告,支持切换分析不同基因
速查表资源
官方文档
- R Markdown速查表
https://rstudio.github.io/cheatsheets/html/rmarkdown.html - Quarto官方文档
https://quarto.org/docs/guide/ - R Markdown权威指南
https://bookdown.org/yihui/rmarkdown/
延伸阅读
- R Markdown Cookbook
https://bookdown.org/yihui/rmarkdown-cookbook/ - Posit全套速查表
https://posit.co/resources/cheatsheets/ - 王敏杰《数据科学中的R语言》
https://bookdown.org/wangminjie/R4DS/
Questions?
王诗翔 副教授
中南大学生物医学信息系
📧 wangshx@csu.edu.cn
🐙 https://github.com/WangLabCSU
下节课预告
- 🔬 上机实验练习
- 📊 撰写你的第一份生信分析报告
谢谢!
![]()
R Markdown基础 | 中南大学